
Dans le monde, il y a déjà eu plus de 664 millions de cas de personnes diagnostiquées avec le COVID depuis le début de l'épidémie qui a commencé en Chine fin 2019 . A cette époque, le virus responsable de la maladie était inconnu, mais depuis, différentes études ont été menées qui ont même permis de suivre son évolution.
En Argentine, des chercheurs scientifiques faisant partie du consortium Proyecto País, du ministère de la Science, de la Technologie et de l'Innovation de la Nation, ont développé un logiciel avec un algorithme qui permet de détecter si une personne a contracté l'infection par plus d'un coronavirus. C'est-à-dire qu'il permet de distinguer si une personne a différentes variantes ou leurs différentes sous-lignées.
Depuis 2020, les chercheurs ont commencé à séquencer les génomes des coronavirus à partir d'échantillons de patients COVID.
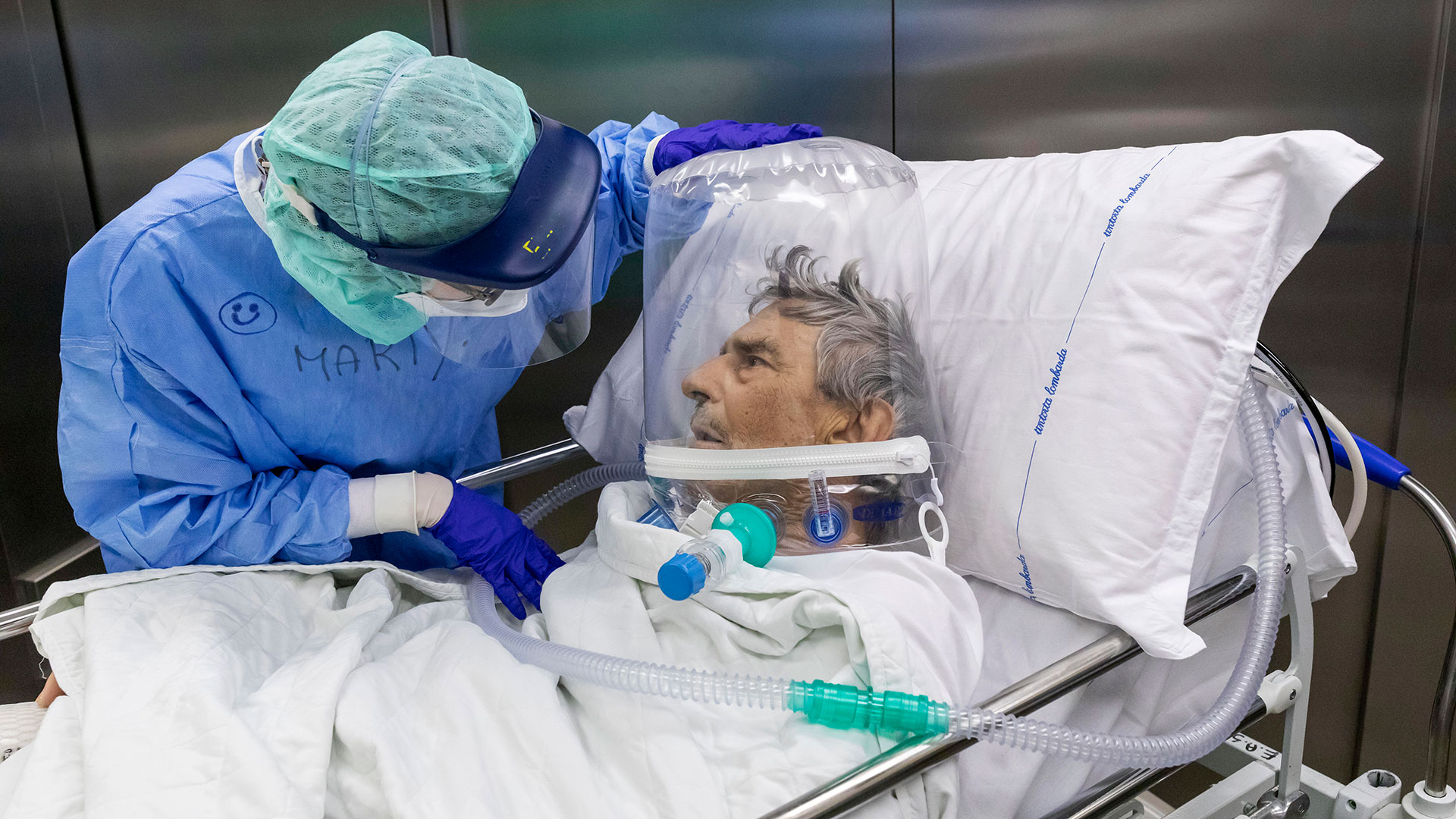
« Nous avons développé un algorithme qui nous permet d'analyser le résultat brut du séquençage par l'une des deux technologies. Il permet de déterminer si plus d'une forme du coronavirus SARS-CoV-2 se trouve dans cet échantillon. Il se pourrait qu'il s'agisse de deux virus de la même lignée, ou bien sûr, de virus de lignées ou de variantes différentes", a déclaré Carolina Torres, co-auteure des travaux et chercheuse en virologie à la Faculté de pharmacie et de biochimie de l'Université de Buenos Aires et l'Université de Buenos Aires, expliqué à Infobae .
La recherche est publiée dans le numéro de février de la revue Virus Research . Dans l'article, l'équipe dirigée par Mariana Viegas, de l'hôpital pour enfants Ricardo Gutiérrez, et Paula Aulicino, du laboratoire de biologie cellulaire et de rétrovirus, qui fait partie de l'hôpital pédiatrique Garrahan, a expliqué que le séquençage génomique du coronavirus s'est avéré être essentiel pour la détection et la réponse à la pandémie.
En faisant ce séquençage, des informations précieuses sont obtenues sur la biologie et l'évolution du virus. Selon la base de données GISAID, plus de 13 millions de séquences complètes du coronavirus ont été générées. Ces informations permettent de suivre en temps réel la diversité génomique du SARS-CoV-2 à travers le monde.
Parmi les différentes technologies de séquençage de nouvelle génération (NGS), Illumina a été la plus couramment adoptée. Représentant 80 % des génomes du SRAS-CoV-2. Bien que les méthodologies de séquençage aient une sensibilité élevée pour détecter plus d'une population virale dans un échantillon, il n'y avait pas de méthode standardisée pour la caractériser.
L'infection simultanée par deux lignées différentes de SARS-CoV-2 a été démontrée pour la première fois peu de temps après le début de la pandémie chez une femme portugaise de 17 ans. Elle était en bonne santé et a développé une maladie grave due au COVID-19.
Il existe également des surinfections par le SRAS-CoV-2, qui sont souvent associées à une excrétion virale prolongée et à des conditions sous-jacentes de l'hôte telles que l'immunosuppression.
Dans les cas de co-infection et de surinfection, l'identification de populations mixtes d'agents pathogènes est plus facile lorsque la divergence virale est élevée. Mais il peut être difficile à détecter dans des contextes de faible hétérogénéité génétique.
"L'identification des co-infections par le SRAS-CoV-2 est plus importante compte tenu de l'apparition de variantes virales avec une évasion immunitaire potentielle et une plus grande transmissibilité, que l'OMS a appelées variantes préoccupantes ou variantes d'intérêt, et la possibilité d'une recombinaison virale entre elles", les chercheurs ont noté dans le document.
Les scientifiques ont ensuite développé "VICOS", qui signifie "Viral Co-Infection Surveillance", consistant en un logiciel open source permettant d'analyser des populations mixtes sur des séquences de coronavirus par la technologie Illumina.
Le logiciel a été utilisé pour effectuer une analyse de 1 097 séquences de SARS-CoV-2 à partir d'échantillons cliniques obtenus entre mars 2020 et août 2021. Des populations mixtes de SARS-CoV-2 ont été trouvées dans 2 % des échantillons. Autrement dit, ils ont détecté 23 cas de personnes atteintes de COVID avec deux virus SARS-COV-2 différents.
Le pourcentage obtenu est similaire à celui attendu selon un petit nombre d'études antérieures qui utilisaient des méthodologies plus complexes pour analyser le comportement évolutif du virus, ont-ils précisé.

La présence de deux virus de la même lignée et très similaire pourrait être une conséquence de l'évolution du SRAS-COV-2 dans l'organisme hôte. Cependant, la présence de virus de lignées différentes montre des événements de coinfection (deux virus infectent en même temps) ou de surinfection (un virus infecte en premier et un autre virus infecte avant la guérison).
En développant le logiciel lVICOS, les chercheurs ont estimé disposer désormais "d'un outil innovant, rapide et très utile pour tous les laboratoires qui réalisent du séquençage génomique pour la surveillance épidémiologique".
Continuer à lire: